I must admit I haven’t got anything to gain from the ‘amyloid hypothesis of Alzheimer’s disease’.
I am not a paid researcher to do with dementia, nor am I paid to promote research campaigns. I do not work for any dementia charities.
In fact, you are entitled to think I am pretty useless. But I am not in fact. I care passionately about dementia research, having done my own work in discovering an innovative way to diagnose the behavioural variant of frontotemporal dementia at Cambridge between 1997-2000. In fact, a key supervisor of my Ph.D. was Prof John Hodges who won a lifetime achievement award at this year’s Alzheimer’s Association conference held in Washington. I consider Hodges a friend as well as colleague. I have been open with him about my personal and professional background. Indeed, John, still knowing all the facts, did me the honour of writing the main Foreword to my first book, “Living well with dementia: the importance of the person and the environment.”
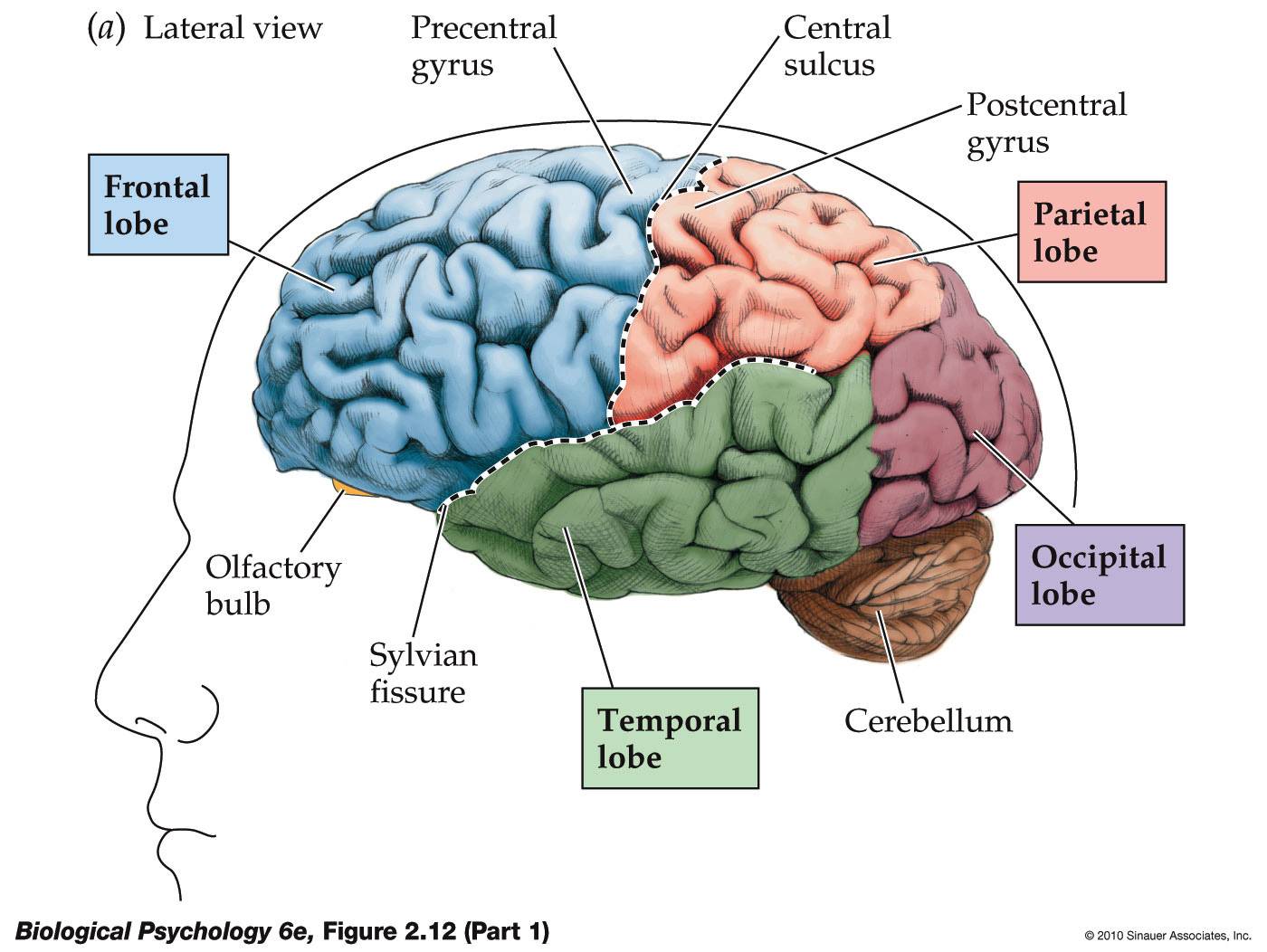
So what is the ‘amyloid hypothesis of Alzheimer’s disease‘? This is, in a nutshell, a build up of an abnormal protein in the brain, as plaques. This build up is like mini brillo pads clogging up the brain, causing the brain to shrink, and buggering up its overall function.
That was my best at being a paid scientific communicator.
But this hypothesis is not without its problems. One key problem is the seminal finding of Price and colleagues (2009) that up to 40% of people without dementia can reach ‘neuropathological criteria’ for Alzheimer’s disease.
And there are in fact perfectly sensible explanations why the amyloid plaques which build up are not the cause of the problems in themselves.
These are summarised well in Morris, Clark and Vissel (2014). It might be that it is type of amyloid plaque which is important to decline; some plaques are non-toxic; amyloid plaques are not actually the cause of Alzheimer’s disease (but soluble beta amyloid might be); and so on.
The amyloid treatments, or else “anti-Aβ treatments’, may have in fact interesting reasons why the “drugs don’t work”.
Back to Morris, Clark and Vissel (2014).
“So far, anti-Aβ treatments have broadly failed to meet their primary clinical endpoints and some major phase 3 trials were halted early. None of the tested treatments have produced a discernible functional recovery, or altered the course of disease. In fact alarmingly some, specifically inhibitors of γ-secretase, lead to an increased decline in cognition.
Another prominent suggested reason for clinical failures of anti-Aβ drugs in particular are that the agents used initially were not properly validated and were flawed. A recent study has shown the monoclonal anti-Aβ antibodies, solanezumab and crenezumab, fail to target human Aβ as effectively as they target over-expressed human Aβ in mouse models. The possibility was also countenanced that only amyloid plaques, potentially functionally inert, rather than soluble Aβ oligomers were targeted in early trials. Furthermore monotherapies may not be capable of effectively reducing Aβ plaque load. A double pronged approach to reduce Aβ by both active immunisation and inhibition of β-secretase has effectively cleared plaques in mice. However, as reviewed recently, therapeutic approaches targeting plaque and approaches targeting soluble Aβ have both now been tested in humans, with equally negative outcomes.”
One idea which had become in vogue was that it was the artist known as ‘fibrillar β-amyloid (Aβ)’ which was the culprit in Alzheimer’s disease. Look at what Stonnington and colleagues reviewed in 2014.
“Fibrillar β-amyloid (Aβ) imaging, most notably with [11C]-benzothiazole radiotracer Pittsburgh compound B (PiB) PET and more recently with the fluorine-18-labeled tracers such as Florbetapir, has emerged as a potential biomarker for preclinical AD. Evidence suggests that increases in fibrillar Aβ deposition precede neuronal injury, and fibrillar amyloid deposition is a potential predictor of later symptomatic cognitive decline. We now have evidence that preclinical cognitive decline correlates with an increased measure of fibrillar Aβ deposition and that this effect is independent of APOE status.”
So we have a bit of a problem on our hands. International scientists, and ‘interested parties’, convening in Washington, for the Alzheimer’s Association International Conference 2015, even if battled were on the whole extremely optimistic. I am not going to call it a trade fair. It is meant to be, a rather, serious discussion of the results, as well as methodological concerns, of rather expensive experiments in the “rolls-royce” end of dementia.
It’s well known that the relationship between the media and reporting of dementia has been in the past a tricky one. Take, for example, this response from the Association of Medical Research Charities, Cancer Research UK and the Wellcome Trust to the ‘Leveson inquiry: Culture, practice and ethics of the press’, dated January 2012.
“Hype and false hope: The flipside of the health scare is the overcooked breakthrough. Many newspapers (though not all of them) are apt to exaggerate interesting but preliminary advances in biomedical science, proclaiming them as groundbreaking achievements that will transform individuals’ health when in fact they are reporting nothing more than promising results from experiments on mice, or cells grown in culture.
Such reporting can have several negative consequences. First, it raises expectations for advances in medical science, many of which will fall by the wayside over the long journey from laboratory bench to patient bedside. This can feed a public perception that science is always promising and never delivering.
Secondly, and more worryingly, it can often raise false hope among patients. This is particularly true and damaging where it concerns treatments for incurable diseases that are not proven, yet which are portrayed as “miracle cures”. This can lead patients to spend life savings on treatments that are most unlikely to work, or on occasion to eschew the most effective known therapies in favour of alternatives that are untested or disproved.”
So we were faced with a difficult situation earlier this week with solanezumab (Lilly).
Key paragraphs from the press release this week are given thus.
“Showing that an investigational treatment has slowed the progression of a degenerative brain disease like Alzheimer’s is extremely challenging. Researchers have proposed overcoming this problem with a type of study called a “delayed-start” trial. In delayed-start studies, patients are randomly assigned to start active treatment at the beginning of the study or are placed in a “delayed-start” group that receives a placebo treatment for a period of time before being given the active experimental therapy. Researchers then compare the two groups at a later, pre-defined point in time to assess their response to the treatment.
…
If the treatment can actually slow disease progression, both groups will benefit, but the group that started active treatment later in the study will have progressed further in the disease before they got the drug – while they were on placebo. As a result, the late starters will not be able to “catch up” to the group whose disease progression was slowed for the full duration of the study.
…
Treatment differences at 28 weeks in EXP-EXT between the early start and delayed start groups for cognition (ADAS-Cog14) and function (ADCS-iADL) were similar to differences at the end of the placebo-controlled period, within a pre-defined margin. In other words, the delayed starters did not “catch up.”
And why 28 weeks?
The answer is actually blindingly obvious – this should be the correct length of time for the drug to have some effect, and our understanding of this is totally dependent on our knowledge of how this drug is metabolised.
Let’s assume that this figure is correct. But even this is not clear-cut.
Turn for a moment to a paper from Liu-Sefert and colleagues this year,
“In a delayed-start design, the placebo-controlled period should be sufficiently long to allow a disease-modifying drug to show an effect and the delayed-start period should be long enough to observe a symptomatic effect. Most clinical studies testing potential disease-modifying treatments have used 18 months for the placebo-controlled period, as in the EXPEDITION studies. Given that the half-life of solanezumab is approximately 28 days, a duration of 28 weeks (approximately 6 months) was chosen for the delayed-start period for the primary efficacy analyses of EXPEDITION-EXT to ensure that the duration of the period was over 5 half-lives and was thus adequate for delayed-start patients to achieve pharmacokinetic equilibrium. The literature also suggests that most symptomatic drugs reach peak effect in less than 6 months. EXPEDITION-EXT is still ongoing and future analyses will demonstrate whether the effect persists beyond 28 weeks.”
Do you remember the fibrillar amyloid which might have an effect?
Well, read for a moment a description of solanezumab thus.
“Solanezumab is a humanized monoclonal IgG1 antibody directed against the mid-domain of the Aβ peptide. It recognizes soluble monomeric, not fibrillar, Aβ. The therapeutic rationale is that it may exert benefit by sequestering Aβ, shifting equilibria between different species of Aβ, and removing small soluble species of Aβ that are directly toxic to synaptic function. In preclinical research, a single injection of m266, the mouse version of solanezumab, reversed memory deficits in APP-transgenic mouse models while leaving amyloid plaques in place, raising the prospect of targeting the soluble pool of Aβ.”
So it doesn’t recognise fibrillar Aβ, but it can reverse memory deficits?
The take-home message there, previously reported only last year, was not good from the phase 3 clinical trial, from Doody and colleagues (Doody et al., 2014):
“Two randomized, double-blind, placebo-controlled, phase 3 studies of solanezumab treatment were performed in patients with mild-to-moderate Alzheimer’s disease. … Solanezumab, a humanized monoclonal antibody that binds amyloid, failed to improve cognition or functional ability.”
Undergraduate students and higher are told that the fundamental problem in Alzheimer’s disease is in the hippocampus, or at least the parahippocampal areas, as this is the heart of learning and memory systems. And yet in the original study the following observation was made (Doody et al., 2014):
“However, the current studies failed to show treatment effects on hippocampal or total brain volumes or on amyloid accumulation with the use of 18 F-florbetapir–PET. These results are consistent with the observation that solanezumab does not target fibrillar amyloid. Our PET findings were not conclusive because of the small sample, but sufficient numbers of solanezumab-treated and untreated patients underwent serial MRI to make the failure to detect a slowing of brain atrophy a meaningful finding.”
The inclusion criteria for the actual trial though is interesting (Doody et al., 2014), and this presumably are the inclusion criteria for the follow up presented at #AAIC2015 this week.
“Both trials involved otherwise healthy patients 55 years of age or older who had mild-to-moderate Alzheimer’s disease without depression. Mild-to-moderate Alzheimer’s disease was documented on the basis of a score of 16 to 26 on the Mini–Mental State Examination (MMSE; score range, 0 to 30, with higher scores indicating better cognitive function) and the criteria of the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association.”
and depression was excluded.
How many clinical raters? If there were more than 1, did they show good agreement? How accurate is a combined score of the MMSE and NINCDS-ADRDA criteria in producing a definitive diagnosis of Alzheimer’s disease? There will be some – and we don’t know how many – who will not have a diagnosis of Alzheimer’s at all in the Alzheimer’s disease randomised patients group.
Vellas and colleagues (2013) had previously warned about a biomarker problem in looking at the methodological issues of solanezumab in doing a ‘lessons learned’ type of report:
“Solanezumab, another humanized monoclonal antibody, targets an epitope in the middle of the Ab peptide. The predominant message emerging from results of the two phase 3 trials (EXPEDITION 1 and 2) is that targeting amyloid appears to have a positive effect on cognition in mild AD, although there were no biomarker changes that indicated a treatment effect.”
As it happens, if the Strictly judges make or break Strictly contestants, it is a concern that investors were not that impressed with the AAIC2015 outcomes. An underwhelming performance of solanezumab was described here this week. Apparently, Lilly shares fell 3.3 percent in early trading.
All of this smacks somewhat of puffery.
I asked somebody who has quite a strong family history of dementia, and a Daily Express reader, what she thought of a possible research development in a cure. She answered straight away, “I think it’s brilliant. Who wouldn’t want a cure for dementia?”
And I have a lot of sympathy with this view. But in law, puffery is a promotional statement which presents subjective rather than objective views. The defense of the media, even reputable broadcasters, that in the relative absence of ‘good news’ about dementia, ignoring completely the vast field of living better with dementia that I and many work in, there is a need to grasp onto any titbits of good news.
But hyperbole is ‘exempt’ from the normal standards of relying on a statement to make a decision.
This is established in English law in a seminal case (albeit only from the Court of Appeal), but the same principles currently apply in other jurisdictions, in the late 19th Century (Carlill v Carbolic Smoke Ball Company 1892).
The United States Federal Trade Commission (FTC) defined puffery as a “term frequently used to denote the exaggerations reasonably to be expected of a seller as to the degree of quality of his product, the truth or falsity of which cannot be precisely determined.”
A “puff piece” is an idiom for “a journalistic form of puffery: an article or story of exaggerating praise that often ignores or downplays opposing viewpoints or evidence to the contrary“.
But there are 47 million people in the world who are relying on news of developments in Alzheimer’s disease and the 100 or so other types of dementia. Some of the Alzheimer’s disease research community have now successfully dug themselves into a hole.
I would strongly urge them to stop digging.
References
Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, Siemers E, Liu-Seifert H, Mohs R; Alzheimer’s Disease Cooperative Study Steering Committee; Solanezumab Study Group.Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014 Jan 23;370(4):311-21.
Liu-Seifert H, Andersen SW, Lipkovich I, Holdridge KC, Siemers E (2015) A Novel Approach to Delayed-Start Analyses for Demonstrating Disease- Modifying Effects in Alzheimer’s Disease. PLoS ONE 10(3): e0119632. doi:10.1371/journal.pone.0119632
Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun. 2014 Sep 18;2:135. doi: 10.1186/s40478-014-0135-5.
Price JL, McKeel DW Jr, Buckles VD, Roe CM, Xiong C, Grundman M, Hansen LA, Petersen RC, Parisi JE, Dickson DW, Smith CD, Davis DG, Schmitt FA, Markesbery WR, Kaye J, Kurlan R, Hulette C, Kurland BF, Higdon R, Kukull W, Morris JC (2009) Neuropathology of nondemented aging: Presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging 30:1026–1036.
Stonnington CM, Chen K, Lee W, Locke DE, Dueck AC, Liu X, Roontiva A, Fleisher AS, Caselli RJ, Reiman EM. Fibrillar amyloid correlates of preclinical cognitive decline. Alzheimers Dement. 2014 Jan;10(1):e1-8. doi: 10.1016/j.jalz.2013.01.009. Epub 2013 Apr 11.
Vellas B, Carrillo MC, Sampaio C, Brashear HR, Siemers E, Hampel H, Schneider LS, Weiner M, Doody R, Khachaturian Z, Cedarbaum J, Grundman M, Broich K, Giacobini E, Dubois B, Sperling R, Wilcock GK, Fox N, Scheltens P, Touchon J, Hendrix S, Andrieu S, Aisen P; EU/US/CTAD Task Force Members. Designing drug trials for Alzheimer’s disease: what we have learned from the release of the phase III antibody trials: a report from the EU/US/CTAD Task Force. Alzheimers Dement. 2013 Jul;9(4):438-44. doi: 10.1016/j.jalz.2013.03.007.
Puffery discussion acknowledgement to Peter Watt.