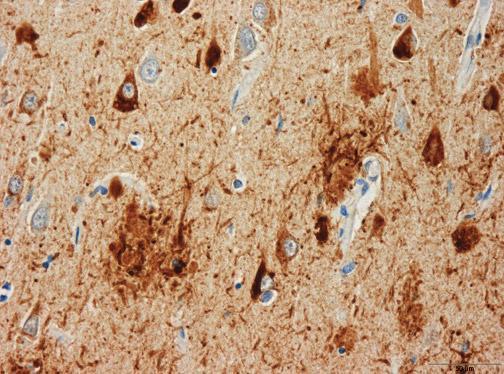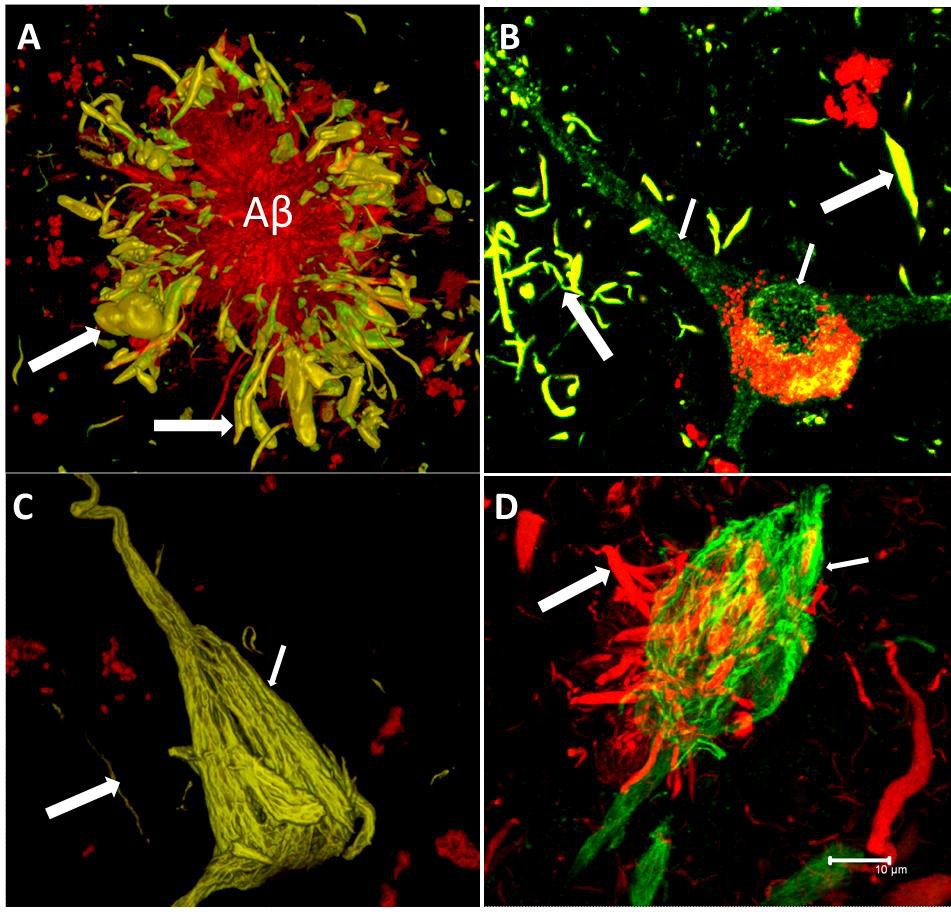
Alzheimer’s disease is the most prevalent type of dementia globally, and therefore traditionally gets the most focus. However, there are other neurodegenerative conditions of note which are of massive importance. For example, neurodegeneration with brain iron accumulation (NBIA) is a group of disorders characterized by dystonia, parkinsonism and spasticity. Models of Alzheimer’s disease, frontotemporal dementia, Parkinson’s disease and Huntington’s disease show some striking similarities to the corresponding human pathologies in terms of axonal transport disruption, protein aggregation, synapse loss and some behavioural phenotypes (Gilley, Adalbert and Coleman, 2011). In early stages of Alzheimer’s disease, neurofibrillary tangles (NFT) are largely restricted to the entorhinal cortex and medial temporal lobe. At later stages, when clinical symptoms generally occur, NFT involve widespread limbic and association cortices. At this point in the disease, amyloid plaques are also abundantly distributed in the cortex.
Induced pluripotent stem cells (also known as iPS cells or iPSCs) are a type of pluripotent stem cell that can be generated directly from adult cells. The iPSC technology was pioneered by Shinya Yamanaka’s lab in Kyoto, Japan, who showed in 2006 that the introduction of four specific genes encoding transcription factors could convert adult cells into pluripotent stem cells. He was awarded the 2012 Nobel Prize along with Sir John Gurdon “for the discovery that mature cells can be reprogrammed to become pluripotent.”
Pluripotent stem cells hold great promise in the field of regenerative medicine. Because they can propagate indefinitely, as well as give rise to every other cell type in the body (such as neurons, heart, pancreatic, and liver cells), they represent a single source of cells that could be used to replace those lost to damage or disease.
According to Wray and Fox (2016),
“It is the use of stem cells as disease models that perhaps has the most to offer in terms of immediate gain, and the most exciting development is the potential to assay potential therapeutics with induced pluripotent stem cells (iPSCs).”
But Wray and Fox (2016) later comment:
“Of particular relevance to Alzheimer’s disease is the fi ending that the expression profile of tau remains feral-like in iPSC-derived neurons until 1 year in culture. Even in cases of familial disease with the earliest onset, the disease only manifests clinically several decades after the onset of pathology and structural changes—how effectively will iPSCs recapitulate the full time course of disease-associated molecular changes?”
Tau proteins (or τ proteins, after the Greek letter by that name) are proteins that stabilise parts of the cell called “microtubules”. They are abundant in neurons of the central nervous system and are less common elsewhere, but are also expressed at very low levels in CNS astrocytes and oligodendrocytes. Pathologies and dementias of the nervous system such as Alzheimer’s disease are associated with tau proteins that have become defective and no longer stabilise microtubules properly.
The tau hypothesis of Alzheimer’s disease states that excessive or abnormal phosphorylation of tau results in the transformation of normal adult tau into PHF-tau (paired helical filament) and NFTs (neurofibrillary tangles). But it is clearly more complicated than that. Deposition of highly phosphorylated tau in the brain is the most significant neuropathological and biochemical characteristic of the group of neurodegenerative disorders termed the tauopathies. Pathological hyperphosphorylation of the microtubule-associated protein tau is characteristic of Alzheimer’s disease and the associated tauopathies. The reciprocal relationship between phosphorylation and O-GlcNAc modification of tau and reductions in O-GlcNAc levels on tau in AD brain offers motivation for the generation of potent and selective inhibitors that can effectively enhance O-GlcNAc in vertebrate brain (Yuzwa et al., 2008).
The discovery of tau fragments in these diseases suggests that tau cleavage and tau phosphorylation, both of which induce conformational changes in tau, could each have roles in disease pathogenesis. As Hanger and Wray (2010) note, the identities of the proteases responsible for degrading tau, resulting in the appearance of truncated tau species in physiological and pathological conditions, are not known.
The Bavarian psychiatrist Alois Alzheimer is traditionally credited with the first description of the most characteristic pathological brain change—neurofibrillary tangles (NFT)—of a yet-unnamed disease in a 51-year-old woman from Frankfurt am Main, who had developed dementia.
During the 1990s, the significance of tau pathology for neurodegenerative diseases, in particular for dementia of the Alzheimer Type, remained in the shadow of the amyloid based approaches. However, as the distribution pattern and overall quantity of amyloid turned out to be of limited significance for pathological staging of AD progression and symptom severity, and after detailed studies of the maturation and distribution of NFTs showing correlation with degree of cognitive decline and memory impairment in Alzheimer’s disease, Braak and Braak proposed a neuropathological staging of the gradual deposition of abnormal tau within vulnerable neurons in brain areas in the form of either NFT or neuropil threads. Post-mortem Braak staging of neurofibrillary tau tangle topographical distribution is one of the core neuropathological criteria for the diagnosis of Alzheimer’s disease.
Based on the biochemically diverse range of pathological tau proteins, Šimić and colleagues (2006) have recently reviewed a number of different approaches which have been proposed to develop new potential therapeutics. Here we discuss some of the most promising ones: inhibition of tau phosphorylation, proteolysis and aggregation, promotion of intra- and extracellular tau clearance, and stabilization of microtubules.
The recent development of candidate PET imaging tracers targeting aggregated tau (now enables not only the brain burden but also the anatomical distribution of tau pathology to be mapped directly in living subjects. One such PET tracer, 18F-AV-1451 (also known as 18F-T807), has been shown to bind selectively to paired-helical filament (PHF) tau, and to exhibit favourable kinetics, low non-specific binding and differential uptake in Alzheimer’s disease versus healthy control subjects. It has been very recently been reported that in vivo 18F-AV-1451 positron emission tomography images across the Alzheimer’s disease spectrum can be classified into patterns similar to those prescribed by Braak neuropathological staging of tau pathology (Schwarz et al., 2016).
But there’s more to tau than Alzheimer’s disease. In NBIA, iron accumulates in the basal ganglia and may be accompanied by Lewy bodies, axonal swellings and hyperphosphorylated tau depending on NBIA subtype (Arber et al., 2015). And there’s more to Alzheimer’s disease than tau. For example, results from Pooler and colleagues (Pooler et al., 2015) strongly support the hypothesis that cortical amyloid accelerates the spread of tangles throughout the cortex and amplifies tangle-associated neural system failure in AD. The story is gradually though unravelling.
Talk
Dr Selina Wray will be giving a presentation at 4 pm today in session 11 of the Alzheimer’s Research UK Research Conference entitled “Modelling tauopathy in patient-derived neutrons: good things come to those who wait?” (link here).
Recommended reading
Arber CE, Li A, Houlden H, Wray S. Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: unifying theories. Neuropathol Appl Neurobiol. 2015 Apr 14. doi: 10.1111/nan.12242. [Epub ahead of print].
Gilley J, Adalbert R, Coleman MP. Modelling early responses to neurodegenerative mutations in mice. Biochem Soc Trans. 2011 Aug;39(4):933-8. doi: 10.1042/BST0390933.
Hanger DP, Wray S. Tau cleavage and tau aggregation in neurodegenerative disease. Biochem Soc Trans. 2010 Aug;38(4):1016-20. doi: 10.1042/BST0381016.
Schwarz AJ, Yu P, Miller BB, Shcherbinin S, Dickson J, Navitsky M, Joshi AD, Devous MD Sr, Mintun MS Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain. 2016 Mar 2. pii: aww023. [Epub ahead of print].
Šimić G, Babić Leko M, Wray S, Harrington C, Delalle I, Jovanov-Milošević N, Bažadona D, Buée L, de Silva R, Di Giovanni G, Wischik C, Hof PR. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules. 2016 Jan 6;6(1). pii: E6. doi: 10.3390/biom6010006.
Wray S, Fox NC. Stem cell therapy for Alzheimer’s disease: hope or hype? Lancet Neurol. 2015 Dec 15. pii: S1474-4422(15)00382-8. doi: 10.1016/S1474-4422(15)00382-8. [Epub ahead of print].
Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ, Vocadlo DJ.29. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol. 2008 Aug;4(8):483-90. doi: 10.1038/nchembio.96. Epub 2008 Jun.